





1-naphthols form an important group of organic molecules. It is a result of their numerous applications, including organic synthesis and pharmaceuticals [1-6]. A lot of them are of very low symmetry, belongs to the C1 point group, there is a chance to find a suitable material for use in organic electronics. Hence there is a need to test as many representatives of this group as it is possible. There have not been published any theoretical studies on 4-chloro-1-naphthol and 4-metoxy-1-naphthol, although their crystal structures had been found [7, 8] and there are *.cif files available for them. This work is a beginning of computational analysis of these two compounds.
Presence of a substituent, which is the hydroxyl group and/or the methoxy group, in the naphthalene aromatic ring allows molecule to exist in several conformational alterations. The aim of this study is to find these isomers and determine energy changes associated with changing orientation of the substituent.
All calculations were made in Gaussian 09 ver. D1 program. The starting point was geometry generated from *.cif file. It was optimized using four different methods (HF, MP2, DFT with the B3LYP hybrid potential and the SVWN local potential) and two basis sets (6-31G(d) and 6-311+G(3d,2p)). Calculations were performed to find stationary points and transition states on the potential energy surfaces, using DFT method with B3LYP hybrid functional in 6-311+G(3d,2p) basis set. 4-methoxy-1-naphthol energies at positions changes of two substituent groups were calculated in 576 points, each geometry was fully optimized.
Structural parameters of free molecules obtained by MP2 and B3LYP methods are in better match to experimental values (measured in the solid phase [7, 8] – crystallographic data from *.cif file) than these from the other type of calculations. Results show, as it often happen, that the larger basis set not always improves results of geometry optimization. After comparison of results of different methods, the quality image of molecules is similar. Differences in lengths of C-C, C-O bonds, do not exceed 0.03 Å and angles do not exceed 1 degree.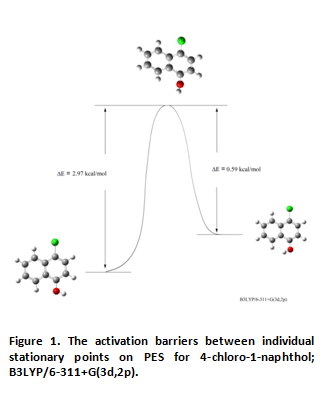
A section through the potential energy surface (PES) of 4-chloro-1-naphthol showed that the compound has two isomers related to changing orientation of the hydroxyl group. In both cases, the hydroxyl group lies in the plane of the ring (as in a case of the 1-naphthol [9]). Energy difference between them is only 1.23 kcal/mole (kT value at 298 K is 0.59 kcal/mole). More stable conformation is the trans isomer, in which the hydroxyl group is directed outside of the molecule, in less stable isomer (cis) OH group is oriented in direction of the second benzene ring. These results correspond to calculations carried out for the unsubstituted 1-naphthol - energy difference between the trans-1-naphthol and the cis-1-naphthol is 1.19 kcal/mole [9]. In the transition state (TS) of 4-chloro-1-naphthol the hydroxyl group is oriented almost perpendicular to the plane of the naphthalene ring. The substituent position change influence in relation to geometry of the molecule remainder is negligible - maximum deviation from the rings planarity is less than 2 degrees.
The scan of potential energy for 4-methoxy-1-naphthol indicates that change in position of the methoxy group requires a considerable energy - activation energy to the transition state is more than 100 kcal/mole. Much more energetically favorable is conformation, in which the methoxy group is facing the second benzene ring. It should be noted that oxygen atom and carbon atom of this group lie in the plane of the naphthalene ring. Changing position of the hydroxyl group (located in the plane of the ring) does not have such a significant impact on energy of the molecule - required energy is approximately 2 kcal/mole. Thus, for this compound there are four possible isomers. At the B3LYP/6-311+G(3d,2p) level there were obtained six optimized structures (three isomers and three transition states). They are given on a figure 2.
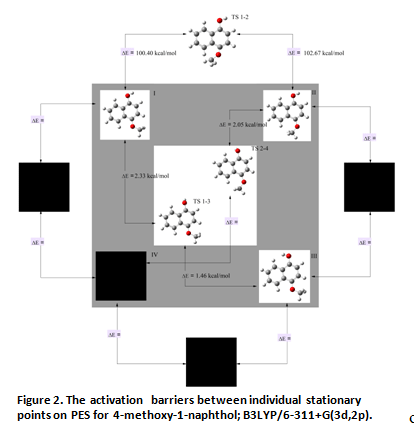
The dark places marked on it correspond to structures, which geometries optimizations were failed with this method. From all isomers, the most energetically preferred is isomer with both groups facing outwards of the ring (designated as I on Figure 1.). Next is III (the hydroxyl group is facing to the second benzene ring), then II (the methoxy group is directed to the second benzene ring, the hydroxyl group to the outside). It can be expected that the least stable is the IV isomer, due to repulsion of charges accumulated on the naphthalene ring atoms and substituent groups atoms lying near this ring.
The analysis of PES obtained with DFT method with B3LYP hybrid functional showed that 4-chloro-1-naphthol and 4-metoxy-1-naphthol exist as conformational isomers. 4-chloro-1-naphthol has two isomers differing in the hydroxyl group orientation. Of these, the trans isomer is more stable. 4-metoxy-1-naphthol has four conformational isomers mutually differing in orientation of the hydroxy and methoxy groups, three of them have been obtained so far. The most energetically preferred is the conformer with both groups facing outwards of the molecule.
References
- X. Huang, G. Zhao, M. Liu, F. Li, J. Qiao oraz S. Zhao, ‘Highly sensitive electrochemical determination of 1-naphthol based on high-index facet SnO2 modified electrode’, Electrochimica Acta (2012), 83, 478– 484.
- Z. Lou, S. Zhang, C. Chen, X. Pang, M. Li, L. Wen, ‘Concise Synthesis of 1-Naphthols under Mild Conditions through a Copper-Catalyzed Arylation of Methyl Ketones’, Adv. Synth. Catal. (2014), 356, 153–159.
- V. Mabuatsela, J. H. Maphoru, K. P. Sreejarani, ‘Oxidative Coupling of 1-Naphthols over Noble and Base Metal Catalysts’, ChemPlusChem (2014), 79, 99 – 106.
- M. El-Agrody Ahmed, M. F. Ahmed, A. E. H. K. Essam Shawky, ‘Synthesis, antitumor activity of 2-amino-4H-benzo[h]chromene derivatives, and structure–activity relationships of the 3- and 4-positions’, Med Chem Res (2013), 22:6105–6120.
- U. Ruktanonchai, O. Nuchuchua, R. Charlermroj, T. Pattarakankul oraz N. Karoonuthaisiri, , ‘Signal amplification of microarray-based immunoassay by optimization of nanoliposome formulations’, Anal. Biochem. (2012), 429, 142–147.
- A. G. Al-Sehemi, A. Irfan oraz A. M. El-Agrody, ‘Synthesis, characterization and DFT study of 4H-benzo[h]chromene derivatives’, Journal of Molecular Structure (2012), 1018, 171–175.
- B. Marciniak, E. Rozycka-Sokolowska, ‘4-Methoxy-1-naphthol: chains formed by O–H···O hydrogen bonds and π– π stacking interactions’, Acta Cryst. (2009), C65, o630–o634.
- E. Rozycka-Sokolowska, B. Marciniak, ‘4-Chloro-1-naphthol’, Acta Cryst. (2005), C65, o207–o210.
- M. Muzomwe, B. Boeckx, G. Maes oraz O. E. Kasende, ‘Matrix isolation FT-IR and theoretical DFT/B3LYP spectrum of 1-Naphthol’, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy (2013), 108, 14–19.