





The study of the structure of phonon spectra has both fundamental and applied significance. It is known that the study of phonon spectra provides important information about the dynamics of the crystal lattice, its structural perfection, allows us to study the phase transformations, etc. Ternary semiconductor crystals of group I-III-VI2 where I = Cu, Ag; III = Al, In, Ga; VI = S, Se, and Te, is a class of materials with chalcopyrite structure. These compounds are important for practical use in various fields, in particular in optoelectronics, photovoltaics, thermoelectricity, nonlinear optics, etc. Recently, the complex theoretical approach for AgGaTe2 crystal study was applied [1]. Also, one of the well-known representatives of this group are AgGaS2 crystals.
The aim of this work is to conduct a theoretical study of the lattice dynamics of ternary chalcogenide AgGaS2 crystals with chalcopyrite structure and to compare the obtained results with the available experimental data to assess the predictive power of the chosen approach.
In this work the first principles calculations within density functional theory (DFT) was used [2-4]. For the description of the exchange-correlation interaction the local density approximation (LDA) was used. The phonon calculation for titled crystal was performed within the density functional perturbation theory, similarly as was done in [1, 5]. For the investigated crystal, the lattice contains N = 8 atoms in the cell. Therefore, the vibrational representation contains 3 × 8 = 24 dispersion branches. Thus, there are 24 normal modes of vibrations in the center of the Brillouin zone in the vibrational spectrum. Three acoustic modes, and the rest are optical. Analysis based on the general method gives the following irreducible representations of acoustic and optical modes in the center of the Brillouin zone: Гvib = 1A1 + 2A2 + 3B1 +4B2 + 7E. The following irreducible representation corresponds to three acoustic modes Гaco = 1B2 + 1E, while the optical vibration has Гopt = 1A1 + 2A2 + 3B1 + 3B2 + 6E representation. The band diagram consists of 24 branches, which is consistent with the number of modes obtained from the symmetric analysis. There is an insignificant dispersion of phonons, which increases when approaching the center of the Brillouin zone. Low-frequency optical branches interact with acoustic ones. The highest frequency of the phonon is 389 cm–1 in the segment between the points Z – Г. The density of states N(ω) indicates two forbidden intervals from 189.4 cm–1 to 219.5 cm–1 and from 255.9 cm–1 to 300.27 cm–1 on which the vibrations occur.
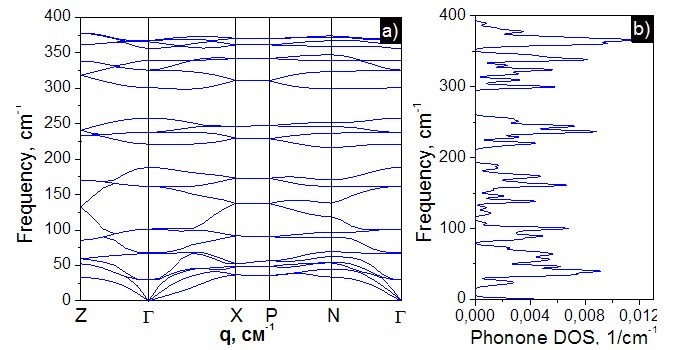
Fig. 1. Calculated phonon dispersion (а) and DOS N(ω) (b) of AgGaS2 crystals.
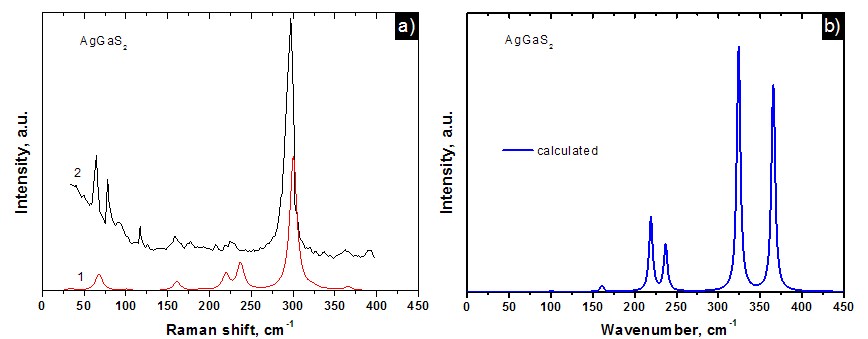
Fig. 2. The Raman (a) and Infrared (b) spectra for AgGaS2 crystal.
The analysis of partial contributions to the phonon density of states is carried out. The Raman and IR spectra are calculated. The experimental Raman spectra reported in [6] have shown a good agreement with the calculated spectra obtained in this study. Obtained results have shown the strong predictive power of theoretical methods in study of the vibrational properties of ternary semiconductors.
M. Ya. Rudysh thanks the support by the PRELUDIUM 15 program of Polish National Science Center (Grant No. 2018/29/N/ST3/02901).
References:
1. M. Ya. Rudysh, M. Piasecki, G.L. Myronchuk, P.A. Shchepanskyi, V. Yo. Stadnyk, O.R. Onufriv, M.G. Brik, Infrared Phys. Technol. 111, 103476 (2020).
2. M. Ya. Rudysh, P.A. Shchepanskyi, A.O. Fedorchuk, M.G. Brik, C.-G. Ma, G.L. Myronchuk, M. Piasecki, J. Alloys Compd. 826, 154232 (2020).
3. A. Majchrowski, M. Chrunik, M. Rudysh, M. Piasecki, K. Ozga, G. Lakshminarayana, I.V. Kityk, J. Mater. Sci. 53 (2), 1217-1226 (2018).
5. R. Muruganantham, W.-R. Liu, C.-H. Lin, M. Rudysh, M. Piasecki, J. Energy Storage. 26, 100915 (2019).
4. M. Ya. Rudysh, A.I. Kashuba, V. Yo. Stadnyk, R.S. Brezvin, P.A. Shchepanskyi, V.M. Gaba, Z.O. Kohut, J. Appl. Spectrosc. 85 (6), 1022-1028 (2019).
6. H. Matsushita, S. Endo, T. Irie, JJAP, 31, 18 (1992).